ゲノム?染色体?DNA?遺伝子?意外と知らない生命科学の基礎知識
理人
リープリーパー


今回は、生物学の歴史において非常に重要なブレイクスルーとなった、ヒト全ゲノムの構築に関する研究を紹介し、そのインパクトを解説します。完全かつ正確な最強のゲノム、その名も「T2T」!
筆者は、ヒトゲノムをはじめとする生命情報を計算機資源で解析するバイオインフォマティシャンで、研究に携わる大学院生です。2022年と2023年のブレイクスルー論文を振り返りながら、実際に普段研究で活用している目線から解説していきます!
「ヒトゲノム」という言葉は聞いたことがあるでしょうか?それは、ヒトの持つ全遺伝情報で、生命の設計図と呼ばれています。その実態は、ACGTの4文字から構成される、約30億文字の長い塩基配列情報です。
実はこの2000年代当初の参照ゲノムのリリースは、未完成のままでした。「タンデムリピート領域」などと呼ばれる配列の繰り返し領域や、染色体の末端領域である「テロメア」部分に関しては、正確に参照配列帰属ができていませんでした。つまり、ヒトゲノムの塩基配列の一部に関しては、技術的な限界からその配列情報を正確に決定できず、一部がモザイク状になっていたのです。
参照配列を決定するプロセスは、パズルを完成させていくようなものです。DNA断片をパズルに見立てた時、それを正確にゲノム全体で辻褄が合うように構築できるか?というものです。ぜひ、この記事も読んでみてください。
「未完成」といっても、「全ゲノムのわずか」が、非常に大きい情報量であることは確かです。直近2019年の最も進んだバージョンの「GRCh38」という参照ゲノムでさえ、約1億5000万塩基対もの未知の配領域が含まれていたようです。現在の遺伝情報学の研究理解では、上記のような不完全ゲノムがきちんと配列決定できていない反復領域が、疾患や遺伝にとって重要である、ということが分かっています。そのため、参照ゲノムを完全に決定することは、生命の理解のために極めて重要なプロセスだったのです。
そして、ついに2年前の2022年にScience誌からブレイクスルーがありました。テロメア・ツー・テロメア(Telomere-to-Telomere;T2T)というコンソーシアムが、「T2T-CHM13」参照配列と呼ばれる実質的に完全なヒトゲノム塩基配列の解読を報告しました。「T2T-CHM13」の公開により、それまでの参照ゲノムで未解明のギャップ領域とされていた領域の、すべての配列が決定されたとのことです。
GRCh38.p13という参照ゲノムと比較すると、T2T-CHM13ではテロメア反復配列(テロメアリピート)が3.6%、縦列反復配列(タンデムリピート)が254%増加しています。参照ゲノムは公開されているので、全世界の研究者は研究資源として活用できる恩恵が得られます。
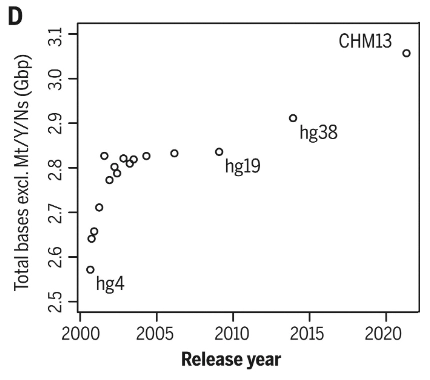
論文にあるグラフを見ると、実際にヒトゲノム計画当初の読まれているゲノムの長さより、2022年のT2T(CHM13)の方がより長いことが分かります。
ヒトゲノムの全長は約30億塩基対であるため、図からT2Tによりほぼ全長が確定したことが読み取れます。
この成功のカギとなったのが、別記事で紹介されている、ロングリード(長鎖)シーケンシング技術でした。「シーケンシング技術」とは、細胞内に実際にあるDNAを読み取ってゲノム配列を決定する技術です。一度に読める配列断片の長さがより長くなるのが、ロングリードシーケシング技術です。
これにより、参照ゲノムの決定精度を妨げる繰り返し配列領域などを、より正確に帰属できるようになりました。これについても、前述のジグソーパズルを例にした記事に詳しく書いているので、一緒にお読みいただけるとわかりやすいでしょう。
参照ゲノム配列を決定するには、ただゲノム断片を読むだけではダメです。実際には、そこからどのように一本の完成したゲノム情報を構築するか、というプロセスは、人が目視で確認できない難しい問題です。そこで、数学的理論に裏付けられた力強いコンピューターアルゴリズムの力が必要です。
アセンブリと呼ばれる手法では、実際に読まれたDNA配列を、グラフ表現と呼ばれる数学的に抽象的な情報フォーマットに落とし込みます。アルゴリズムを解くことで、より正確かつ完全な参照ゲノムを構築できるようになりました。
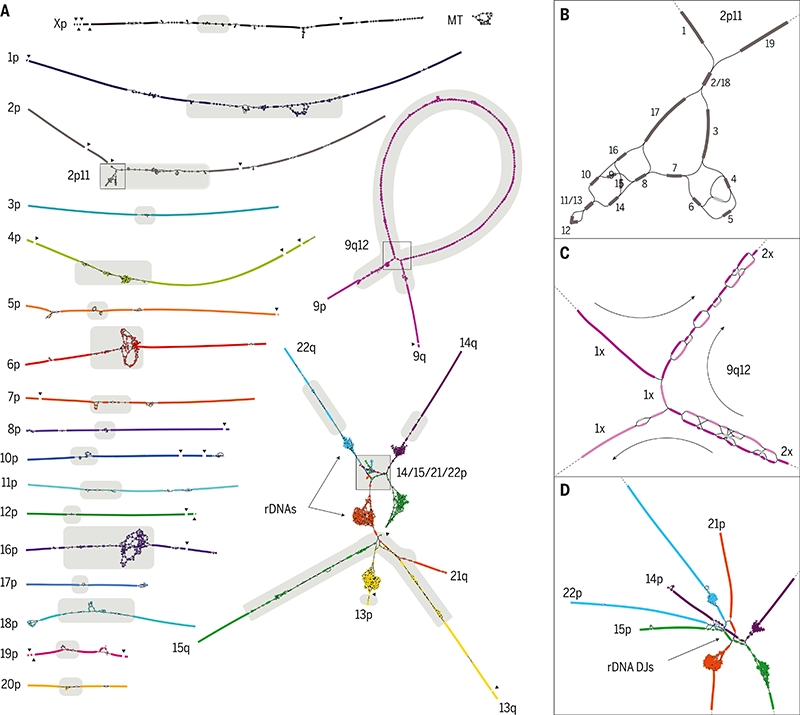
T2T-CHM13参照配列によって、ヒトの平均的な参照ゲノムが構築されました。これには、反復領域など、今まで遺伝子の暗黒領域「ダークマター」と呼ばれる、よく分からないもやもやした部分までを含んでいます。しかし、これはまだ序章に過ぎず、次にはヒト一人あたりの完全な遺伝的多様性を捉えた、完全な個人遺伝情報を構築できる未来が来ます。
基本的にヒト一人ひとりが持つ遺伝情報は、参照ゲノムと比べて微妙に異なっています、いわゆる遺伝子に変異が起きているのです。人々の間には遺伝学的な違いがあるため、T2Tゲノムと比較的よく似た配列のゲノムを持つ人もいれば、違った箇所の多いゲノムを持つ人もいるのです。
そして、これら遺伝的な多様性が、遺伝性疾患や遺伝形質、疾患感受性の根底にあることは、セントラルドグマの原理を振り返れば明らかです。新しいブレイクスルーは、恐らく1年間隔でどんどん更新されていきます。最高のアルゴリズムと解析技術の到来と、時代の変化を肌で感じていきましょう。
私たちは、自身を構築する完全かつ究極の個人情報を把握できる時代に生きています。そして、大規模言語モデルや医療ビッグデータセンター、IoTなど、既に我々人類が構築してきた情報インフラを通じて、有効活用されていくのです。これは、別記事でも述べた ”Bio is new Digital” 的な世界観を加速していくことを意味しています。わくわくしますね!